Research | Open Access | Volume 9 (Suppl 12): Article 04 | Published: 24 Apr 2026
Views: 479
Menu, Tables and Figures
| Table 1: Docking and MMGBSA Scores for the Top Drug Candidates | ||
|---|---|---|
| Drug candidate | MMGBSA dG Bind (kcal/mol) | Docking score (kcal/mol) |
| Chloroquine | -40.66 | -4.109 |
| 3-hydroxyquinine | -36.95 | -4.01 |
| Tetracycline | -36.69 | -6.494 |
| Artesunate | -35.17 | -4.4 |
| Chlorproguanil | -34.18 | -4.021 |
| Doxycycline | -30.27 | -4.796 |
| Monodesethylamodiaquine | -29.33 | -4.115 |
| Proguanil | -29.23 | -4.022 |
| Clindamycin | -27.78 | -5.828 |
| Co-ligand | -12.02 | -6.191 |
Table 1: Docking and MMGBSA Scores for the top drug candidates
| Table 2: Post docking analysis showing the H bond, Hydrophobic interactions, and other interactions for the top six drug candidates | |||
|---|---|---|---|
| Drug candidate | H bond | Hydrophobic interactions | Other interactions |
| Chloroquine | None | CYS 709, TRP 795, ILE 797, ALA 799, TYR 766, TRP 803, MET 765, MET 761, LEU 511 | Pi-pi stacking: HIS 711 |
| 3-hydroxyquinine | ARG 729, SER 796 | LEU 734, TYR 758, TYR 766, MET 761, TRP 795, LEU 511, ALA 799, CYS 709 | Pi-pi stacking: HIS 711 (2) |
| Tetracycline | ARG 729, CYS 709, SER 796 (2), ASP 664, SER 661 | MET 761, TYR 766, LEU 511, CYS 709, ILE 797, ALA 799, TYR 606 | None |
| Artesunate | TYR 766, TRP 795 | TRP 803, TYR 758, MET 761, MET 765, TYR 766, LEU 511, TRP 795, LEU 734 | None |
| Chlorproguanil | SER 710 | TRP 803, MET 761, TYR 766, LEU 511, ILE 797, ALA 799, CYS 709, TYR 606 | Pi-pi stacking: HIS 711 |
| Doxycycline | SER 661, SER 796 | TYR 766, MET 761, LEU 511, TRP 795, ILE 797, ALA 799, CYS 709, TYR 606 | None |
Table 2: Post docking analysis showing the H bond, Hydrophobic interactions, other interactions for the top six drug candidates
Adeniyi Ayinde Abdulwahab1,&, Olumide Oluyele2, Emmanuel Ayomide Adedibu3
1Faculty of Pharmaceutical Sciences, Bayero University, Kano, Nigeria, 2Department of Microbiology, Adekunle Ajasin University, Akungba-Akoko, Nigeria, 3Department of Microbiology, Faculty of Life Sciences, University of Ilorin, PMB 1515, Ilorin, Kwara State, Nigeria
&Corresponding author: Adeniyi Ayinde Abdulwahab, Faculty of Pharmaceutical Sciences, Bayero University, Kano, Nigeria, Email: adeniyiabdulwahab3@gmail.com ORCID: https://orcid.org/0000-0002-0940-0014
Received: 09 Nov 2025, Accepted: 23 Apr 2026, Published: 24 Apr 2026
Domain: Infectious Disease Epidemiology
Keywords: Drug repurposing, anti-malarial drug, dengue virus, molecular docking, MMGBSA
©Adeniyi Ayinde Abdulwahab et al. Journal of Interventional Epidemiology and Public Health (ISSN: 2664-2824). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Adeniyi Ayinde Abdulwahab et al., Structure-based repurposing of antimalarial drugs to inhibit the RNA-dependent RNA polymerase of dengue virus. Journal of Interventional Epidemiology and Public Health. 2026; 9(Suppl 12):04. https://doi.org/10.37432/jieph-d-25-00278
Introduction: Dengue fever is becoming a global health emergency, being the most widespread mosquito-borne viral disease, putting half the world population at risk of infection. Dengue virus (DENV), the causative agent of the disease, is classified into four serotypes (DENV-1 – DENV-4), each associated with fever and dengue shock syndrome. Currently, no antiviral drugs are approved for the disease; treatments are based on supportive care. This study follows a comprehensive structure-based virtual screening approach to screen a collection of approved antimalarial drugs for binding efficiency and inhibitory potential against DENV RNA-dependent RNA polymerase (DENV RdRp).
Method: We retrieved thirty-one (31) approved antimalarial drugs from published literature. This was followed by downloading the three-dimensional structures (3D) of each drug from the PubChem website and the crystal structure of the protein (PDB ID: 2J7W) from the Protein Data Bank. We computed the root mean square deviation (RMSD) to validate the docking study. The molecular docking and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) evaluation were performed using the Maestro Schrodinger software user interface. The Maestro’s molecular visualisation tool was utilised to perform a post-docking analysis for each of the top drug candidates
Result: The computed RMSD for the redocked cocrystallized ligand was 2.0 Å. The docking scores for the top 5 drug candidates were Chloroquine (-4.109 kcal/mol), 3-hydroxyquinine (-4.01 kcal/mol), Tetracycline (-6.494 kcal/mol), Artesunate (-4.4 kcal/mol) and Chlorproguanil (-4.021 kcal/mol). The MMGBSA (dG bind) for the top five drug candidates were Chloroquine (-40.66 kcal/mol), 3-hydroxyquinine (-36.95 kcal/mol), Tetracycline (-36.69 kcal/mol), Artesunate (-35.17 kcal/mol), and Chlorproguanil (-34.18 kcal/mol). The post-docking analysis revealed considerable intermolecular interactions between the drug candidates and protein.
Conclusion: Several clinically approved antimalarial agents, including chloroquine, 3-hydroxyquinine, tetracycline, and artesunate, demonstrated favourable binding affinities and stable interactions with catalytically essential residues of the enzyme (DENV RdRp). These interactions suggest potential inhibitory effects on viral replication consistent with previous in vitro and in vivo observations. Future studies should integrate molecular dynamics simulations, enzymatic inhibition assays, and animal model testing to confirm their antiviral efficacy and clarify the molecular basis of NS5 inhibition.
Dengue fever is becoming a global health emergency, as it is the most widespread mosquito-borne viral disease, putting half the world’s population at risk of infection. Dengue cases have been reported across 100 countries in both tropical and subtropical regions. The disease continues to expand its geographical spread. Recent epidemiology data show the disease affects 100-400 million people annually, with approximately 96 million clinically severe cases, many of which go unreported [1,2]. The world recorded the worst dengue outbreak in the last two years; in 2023, 6.5 million cases were diagnosed, which climbed significantly to 14 million in the following year, resulting in approximately 9,000 deaths [2,3,4]. Another source reported 12 million cases and 7,152 deaths in the Americas by mid-2024 [3]. Several factors favour this unprecedented rise in dengue cases, such as climate change, increased global travel, and the expanding range of Aedes mosquito vectors [1, 5,6].
Dengue virus (DENV), the causative agent of the disease, is a member of the Flaviviridae family and classified into four serotypes (DENV-1 – DENV-4), each associated with fever and dengue shock syndrome [7]. Although lifelong immunity has been observed in survivors of one serotype [8], infection with a sister serotype poses a severe risk of complications through antibody-dependent enhancement, the same phenomenon associated with vaccine usage [9,10]. Currently, no antiviral drugs are approved for the disease; treatments are based on supportive care [1,11]. ‘Dengvaxia,’ which is the current vaccine, is exclusive to individuals within the 6-16-year-old age group who have clinically confirmed previous cases to lower the risk of complications when exposed [12,13]. To further address this, Takeda has an ongoing project of another vaccine, ‘TAK-003,’ (Qdenga), a live-attenuated vaccine comprising all four serotypes.
The DENV non-structural protein 5 (NS5), the largest and most evolutionarily conserved protein of flaviviruses, is a prime target in antiviral drug development projects. It comprises an N-terminal methyltransferase (MTase) domain and a C-terminal RNA-dependent RNA polymerase (RdRp) domain overseeing its RNA replication all in its 900 amino acid stretch [14,15,16]. The enzyme has a pronounced right-handed structure with a highly conserved active site not present in mammalian hosts, positioning it as a viable target for the design of broad-spectrum antivirals [16]. However, only a few RdRp inhibitors have made it to clinical [17,18]. High-throughput screening techniques have been further employed to make headway and have identified candidate chemical scaffolds with DENV inhibitory activity in vitro [19]; however, clinical progress of these candidates is hampered by potency, selectivity, pharmacokinetics, and serotype coverage concerns [20,21,22]. The absence of DENV-specific approved antivirals raises an urgent need for innovative drug discovery methods.
Drug repurposing, or drug repositioning, is a promising accelerated strategy for therapeutic discovery by searching for alternative uses for existing drugs [23,24]. Apart from being a faster and low-cost approach, repurposing re-evaluates drugs with established safety profiles, compared to entirely new compounds [25]. It could facilitate immediate medical relief during emergency health threats such as the recent COVID-19 pandemic [24]. Earlier reports have reported the antiviral potentials of antimalarial drugs, making them promising candidates for viral inhibition, including DENV [26,27,28].
Drug repurposing could be particularly useful during emergency health situations, especially when novel pathogens or strains are involved, which require immediate response, such as the recent COVID-19 pandemic [24]. Antimalarial drugs have also been shown to have antiviral potential and could be viable candidates for viral inhibition [26,27,28]. Structure-based drug design (SBDD), a focal point of computer-aided drug design, enables the simulation of drug interactions with target enzymes, thus offering a faster and cost-effective approach in the search for new therapeutics [29,30]. Applied to drug repurposing for antiviral functions, it complements ongoing efforts by rapidly highlighting promising candidates for experimental validation.
This study follows a comprehensive structure-based virtual screening approach to screen a collection of approved antimalarial drugs for binding efficiency and inhibitory potential against DENV NS5 RdRp. We assessed their binding energy at the enzymatic active site and pharmacokinetic properties to identify lead compounds for further in vitro and in vivo validation towards developing dengue therapeutics.
Ligand selection and preparation
We retrieved thirty-one (31) approved antimalarial drugs from published literature. This was followed by downloading the three-dimensional structures (3D) of each drug from the PubChem website and importing them into the Maestro Schrödinger software. Furthermore, we employed the LigPrep module of Maestro Schrödinger to prepare each drug utilizing the OPLS3 force field at a target pH of 7.0 to ensure that the drugs conform to the molecular states required for the molecular docking study.
Protein preparation
The crystal structure of the target receptor, Dengue virus NS5 RNA-dependent RNA polymerase domain complexed with 3′ dGTP (PDB ID: 2J7W) was obtained from the Protein Data Bank (https://www.rcsb.org/structure/2J7W ) [31]. The protein was imported into Maestro workspace and subsequently preprocessed using the Protein Preparation Wizard by removing the di(hydroxylethyl) ether, zinc ion, and water molecules. The protein was optimized by adding hydrogen bonds using PROPKA at pH 7.5, and later minimized utilizing the OPLS3e force field.
Receptor grid generation and docking validation
A receptor grid was generated from the minimized protein through the Glide grid module to specify the active site of Dengue Virus RNA-Dependent RNA Polymerase (DENV RdRp) catalytic domain for ligand-receptor docking. This was achieved by selecting the cocrystalized ligand (guanosine-5′-triphosphate) at the active site of the protein to produce a cubic grid box with three-dimensional coordinates X, Y, and Z having values of 18.58, 67.06, and 14.29, respectively. To ensure the reliability of the molecular docking study, the co-crystalized ligand was redocked into the active site of DENV RdRp to ensure the root mean square deviation (RMSD) is within the acceptable range.
Molecular docking and post-docking analysis
We utilised the Glide module within the Maestro interface of Schrödinger 11.8 to conduct an extra precision (XP) molecular docking study under the following conditions: default scaling factor of 0.8, and partial charge cutoff of 0.15. However, we left the default setting unchanged, including samples, nitrogen inversions, sample ring conformations, and the addition of Epik state penalties to the docking score. Following the docking study, we utilised the Maestro’s molecular visualisation tool to perform a post-docking analysis for each of the top drug candidates to evaluate the protein-ligand interaction with focus on the following key parameters: hydrogen bonds, hydrophobic interactions, and other interactions (pi-pi stacking). This analysis gives a clear overview of the binding poses of each drug candidate within the active site of the protein and the intermolecular interactions that took place.
Estimation of binding energy using MMGBSA
We estimated the free binding energy using Molecular Mechanics-Generalised Born Surface Area (MM-GBSA) through Maestro’s Prime module to further validate the potential of the top ligands from the XP molecular docking study as suitable candidates against the DENV RdRp. Only the top five drug candidates from the XP molecular docking analysis were employed in this study, using a default OPLS3 force field and the VSGB solvation model.
Docking protocol validation
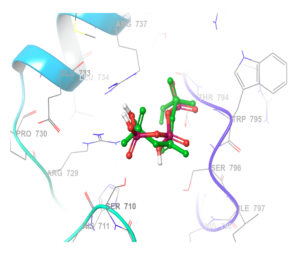
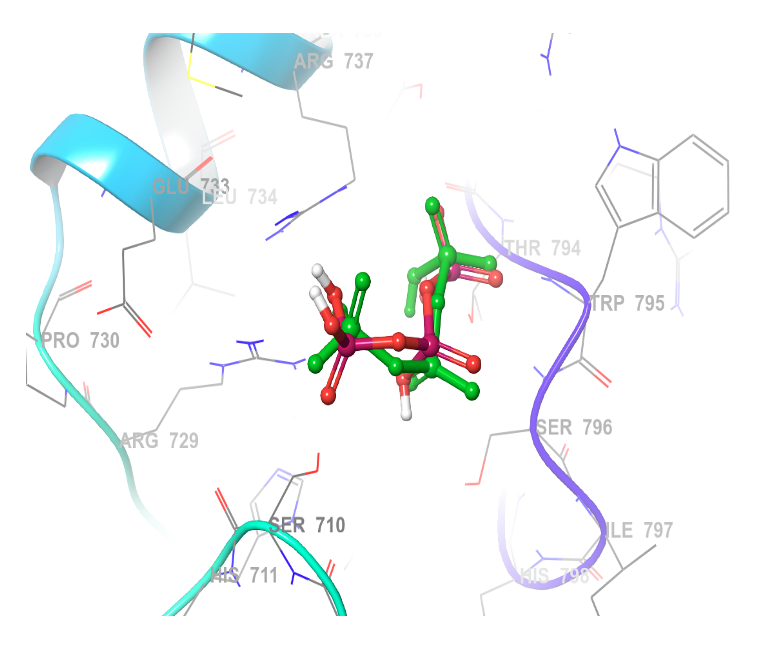
To ensure the reliability of the molecular docking approach, the protocol was first validated by redocking the cocrystallized ligand into the active site of the dengue virus (DENV RdRp) (PDB: 2J7W). The redocked ligand had a root mean square deviation (RMSD) of 2.0 Å, in reference to the native ligand, which falls within the accepted threshold (≤2.0 Å), confirming the accuracy of our docking protocol for subsequent studies [32]. The superimposed structure of the redocked cocrystallized ligand on the native ligand (Figure 1).
Virtual screening and molecular docking of antimalarial compounds
The native position of the co-crystalised ligand was used to determine the enzymatic active site of DENV RdRp ligand docking. The grid box is centered at coordinates X: 18.58, Y: 67.06, Z: 14.29. This region encompasses critical residues responsible for its enzymatic function and a useful binding site for inhibitors. Figure 1 shows the co-crystallised ligand interaction with the enzyme at its site.
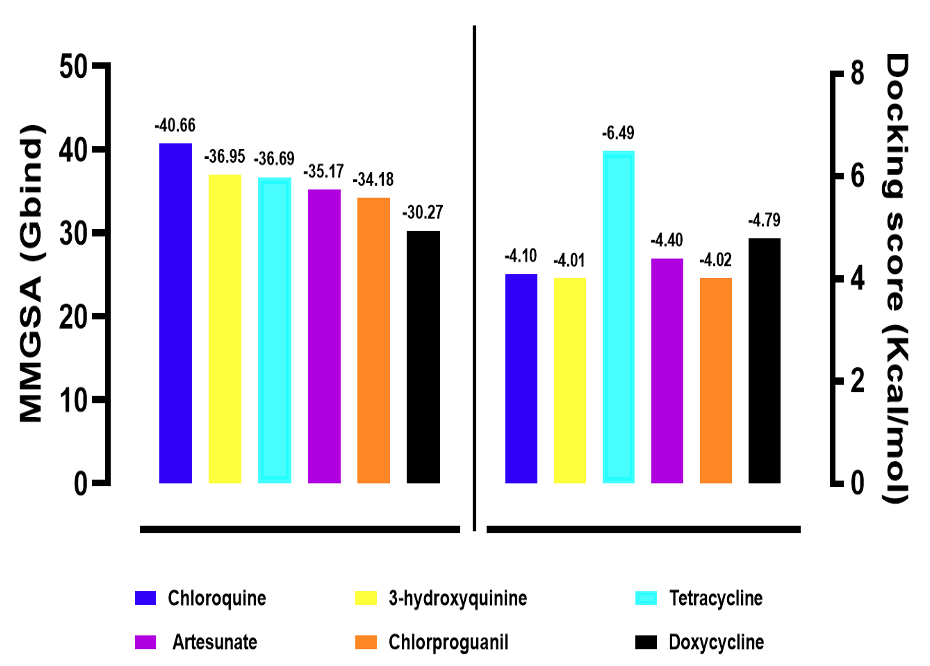
A library of 30 antimalarial drugs was retrieved and docked against the defined active site of DENV RdRp using the Schrödinger suite in extra precision (XP) mode. The docking workflow further evaluated the binding free energies (MMGBSA dG Bind) to estimate ligand affinity. Docking and MMGBSA metrics revealed several antimalarial drugs with promising binding affinities for the DENV RdRp active site (Table 1, Figure 2). The five compounds with the most favourable MMGBSA binding energies were chloroquine, 3-hydroxyquinine, tetracycline, artesunate, and chlorproguanil. Tetracycline exhibited the strongest docking score (-6.494 kcal/mol) and substantial binding free energy (-36.69 kcal/mol). Chloroquine showed the most favourable MMGBSA binding free energy (-40.66 kcal/mol), followed by 3-hydroxyquinine, artesunate, and chlorproguanil, having -36.95 kcal/mol, -35.17 kcal/mol, and -34.18 kcal/mol, respectively. For comparison, the co-crystallised ligand (reference inhibitor) yielded a MMGBSA dG Bind of -12.02 kcal/mol and a docking score of -6.191 kcal/mol.
Post-docking interaction analysis
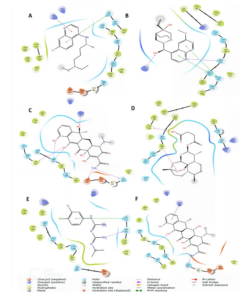
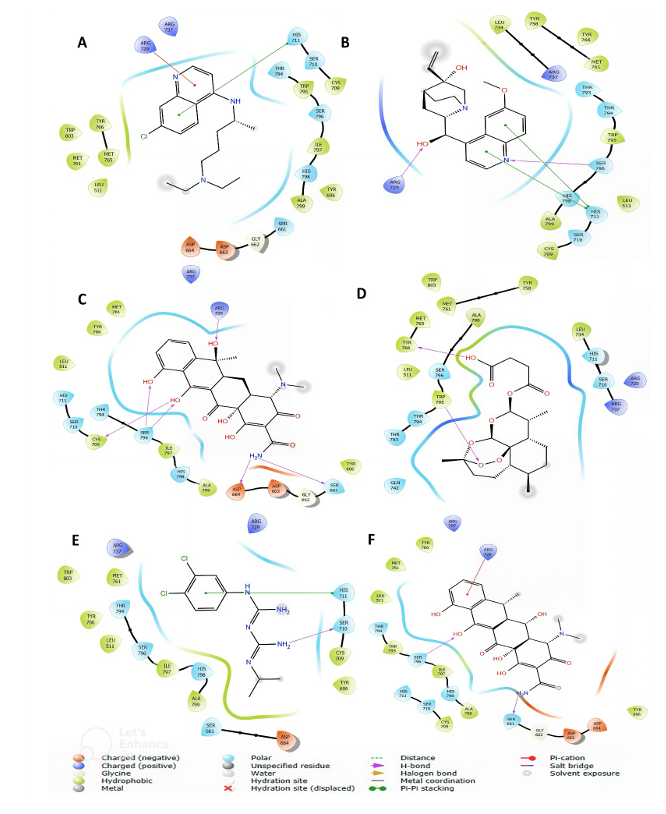
Post-docking analysis gave details of molecular interactions between the top six antimalarial drug candidates and the DENV RdRp active site (Table 2), which varied across the compounds. Chloroquine showed no hydrogen bonds but formed extensive hydrophobic interactions with nine active site residues and a key pi-pi stacking with HIS711. 3-hydroxyquinine has two hydrogen bonds with ARG729 and SER796 residues of the active site, along with hydrophobic contacts and dual pi-pi stacking at HIS711. Tetracycline showed more extensive interaction with the enzymatically active site by establishing multiple hydrogen bonds with both backbone and side-chain atoms and broad hydrophobic interactions. Artesunate had two hydrogen bonds (TYR766, TRP795) with several hydrophobic interactions, likewise chlorproguanil and doxycycline. Only chloroquine, 3-hydroxyquinine, and chlorproguanil have Pi-pi stacking interactions with HIS 711. Figure 3 gives visual highlights of the binding interactions of the top candidates at the DENV RdRp active site, which account for ligand affinity and specificity.
Dengue virus (DENV) is a mosquito-borne flavivirus responsible for dengue fever and its severe manifestations, including dengue hemorrhagic fever and dengue shock syndrome, which continue to pose significant global health challenges in tropical and subtropical regions [33]. The virus comprises four antigenically distinct but closely related serotypes (DENV-1 to DENV-4) that share high structural and genetic similarity [34], complicating both vaccine development and antiviral therapy. Despite extensive research, no specific antiviral drug has been approved for dengue treatment, highlighting the need to identify novel molecular targets and explore drug repurposing strategies.
The dengue virus non-structural protein 5 (NS5) is a multifunctional enzyme responsible for both RNA capping and RNA-dependent RNA polymerase (RdRp) activities, which are essential for viral replication and immune evasion [17,35]. Its highly conserved structure among flaviviruses makes it an attractive target for antiviral drug development and repurposing. The PDB entry 2J7W represents the RdRp domain of DENV serotype 3 complexed with 3′-deoxyguanosine triphosphate (3′ dGTP) [31], a nucleotide analog that binds to the polymerase active site and serves as a reference for evaluating inhibitor interactions.
In-silico drug discovery techniques such as molecular docking and MM/GBSA binding free-energy calculations have become indispensable in modern antiviral research due to their ability to predict ligand–target interactions, guide hit prioritization, and minimize experimental costs [36,37]. While molecular docking provides a rapid assessment of potential binding orientations and affinities, MM/GBSA calculations offer more refined thermodynamic estimates by combining molecular mechanics energies with solvation and surface area terms [38,39]. These computational approaches collectively improve the accuracy of virtual screening outcomes and strengthen the reliability of predictions prior to in vitro validation.
In this study, molecular docking and MM/GBSA analyses were employed to evaluate the binding interactions of selected antimalarial drugs with the DENV NS5 polymerase (PDB ID: 2J7W). The MM/GBSA results provided a semi-quantitative assessment of ligand stability within the active site, accounting for solvation effects and partial receptor flexibility often neglected in rigid docking. Among the screened compounds, chloroquine exhibited the most favourable dG_bind (–40.66 kcal/mol), followed by 3-hydroxyquinine (–36.95 kcal/mol), tetracycline (–36.69 kcal/mol), and artesunate (–35.17 kcal/mol). These values were substantially lower than that of the co-ligand (–12.02 kcal/mol), suggesting stronger predicted inhibition within the NS5 active site. The RMSD value of 2.0523 Å for the redocked co-ligand further validated the docking accuracy and reliability of the computational protocol.
It is important to note that the docking scores and MM/GBSA binding energies did not exhibit perfect numerical correlation. This divergence is expected because the two methods evaluate binding energetics using fundamentally different models. Docking scores are generated from static ligand–protein poses using simplified empirical scoring functions that emphasize shape complementarity and local interactions. In contrast, MM/GBSA incorporates molecular mechanics energies and solvation contributions across multiple conformations, capturing aspects of induced fit and dynamic stability that docking does not model. As a result, complete correlation between the two outputs is not anticipated. Instead, their complementary nature allows more confident prioritization of ligands when compounds consistently perform well across both analyses.
The differences in scoring also highlight the importance of using multiple computational approaches in virtual screening. While docking provides a rapid means to rank large compound libraries, MM/GBSA offers a more physically realistic estimate of binding free energy by including solvent effects and partial protein flexibility. Taken together, the combined application of these methods strengthens confidence in the identified top candidates and informs the selection of compounds for subsequent experimental validation.
The interaction profiles revealed that the top-scoring compounds established multiple hydrophobic and hydrogen-bond contacts with catalytically important residues, including TYR766, TRP795, MET761, and CYS709, which stabilize nucleotide binding and enzyme catalysis. The π–π stacking between HIS711 and the aromatic rings of chloroquine, chlorproguanil, and 3-hydroxyquinine may further reinforce binding stability, highlighting their potential as lead scaffolds for antiviral repurposing. Moreover, per-residue energy decomposition is a capability of MM/GBSA that can provide mechanistic insight into individual residue contributions and help identify “hot spots” for inhibitor design [40]. In this study, mechanistic insights were inferred from the observed docking and MM/GBSA interactions rather than from explicit per-residue decomposition.
The findings of this study align with an expanding body of computational research identifying the DENV NS5 polymerase as a highly tractable antiviral target. Earlier experimental evidence already supports the antiviral potential of several antimalarial agents investigated here. Chloroquine and related quinoline derivatives have been shown to inhibit DENV replication by blocking endosomal acidification and interfering with viral RNA synthesis [41,42]. Artesunate and other artemisinin derivatives similarly suppress viral propagation through modulation of oxidative stress and host immune pathways [43], while doxycycline exhibits antiviral properties by inhibiting viral protease activity and attenuating pro-inflammatory cytokine responses [44,45]. These mechanistic reports provide biological plausibility for the strong NS5 binding interactions observed in the present computational analysis, reinforcing the feasibility of repurposing existing antimalarial agents as potential DENV polymerase inhibitors.
Virtual-screening studies further corroborate the trends observed in this work. Galiano et al. (2016) conducted one of the earliest large-scale structure-based screens of the NS5 RdRp, evaluating 372,792 non-nucleotide compounds using AutoDock/Vina and identifying 39 promising inhibitors with binding energies below –10.5 kcal/mol after ADMET refinement [46]. Despite differences in compound libraries (natural-product-rich chemical space vs. clinically approved antimalarials) and docking platforms (AutoDock/Vina vs. Schrödinger Maestro), their results converge with ours in highlighting the RNA-template tunnel and surrounding catalytic residues as highly responsive to small-molecule inhibition. Notably, the binding free energies of chloroquine, 3-hydroxyquinine, artesunate, and doxycycline in our study fall within the range reported for several high-affinity ligands identified by Galiano et al., demonstrating that distinct chemical classes can achieve comparable inhibitory profiles against NS5.
More recent computational work reinforces this convergence. Hasan et al. (2025) screened 270 phytochemicals from edible vegetables and found apigenin-7-glucoside, rutin, and phlorizin to exhibit superior docking scores (–8.8 to –8.1 kcal/mol) relative to the reference antiviral favipiravir (–7.4 kcal/mol) [47]. Their top hits formed extensive hydrogen-bonding and hydrophobic interactions with active-site residues such as LYS355, GLY599, PHE354, and VAL358. MM/PBSA analysis further confirmed their favourable binding energetics throughout MD trajectories. Although Hasan et al. employed AutoDock, the overarching outcomes—strong phytochemical affinity, stable binding, and energetically favourable interactions—mirror the patterns observed in our MM/GBSA results, further validating the robustness of NS5 as a druggable target across docking engines [47].
Additional support comes from the large-scale natural-product screening conducted by Putra et al. (2020), who evaluated 343,798 ZINC15 compounds against NS5 RdRp structures from all four DENV serotypes [46]. Their top leads—Compounds 3556, 9487, and 106665—demonstrated strong inhibitory potential, with Compound 3556 achieving a notably favourable binding energy (dG_binding = –11.1618 kcal/mol) through optimal interactions with catalytic residues. The consistency between their findings and ours underscores the versatility of NS5 as a molecular target capable of accommodating structurally diverse inhibitors, including both natural products and repurposed antimalarial drugs.
Collectively, the literature consistently demonstrates that the DENV NS5 RdRp is highly amenable to small-molecule inhibition and that diverse compound classes—including antimalarial agents, phytochemicals, and synthetic non-nucleotides—can stably occupy functionally important regions of the polymerase. The strong docking and MM/GBSA binding energies observed in this study, particularly for chloroquine, 3-hydroxyquinine, tetracycline, and artesunate, are therefore well supported by prior work and further highlight the potential of these scaffolds for antiviral drug development. From a drug development standpoint, repurposing antimalarial agents against DENV offers notable advantages, including well-characterised pharmacokinetic and safety profiles, as well as accessibility in co-endemic regions. The structural resemblance of quinoline and tetracycline scaffolds to known nucleoside analogue inhibitors [48,49,50] may explain their favourable interactions with viral polymerases, emphasising the need for further experimental validation in relevant infection models.
Limitations of the study
This study has important limitations that should be acknowledged. First, the co-ligand retrieved from the PDB structure (2J7W) differs from the original cocrystallized 3′-dGTP molecule. The available ligand corresponds only to the phosphate-chain moiety of the nucleotide, which, although sufficient for identifying key active-site residues and calculating RMSD, is not an ideal reference for comparative binding evaluation. This structural incompleteness may influence the accuracy of binding orientation and affinity assessments. Future docking studies should utilise full ligand structures or validated reference inhibitors to enhance reliability.
Second, the workflow employed in this study is entirely computational. While in silico screening provides a robust and cost-effective approach for preliminary prioritisation of candidate molecules, the absence of complementary in vitro or in vivo validation limits the direct biological interpretation of the predicted inhibitory activities. To address this, future investigations will incorporate cell-based assays and animal models to experimentally confirm the efficacy, pharmacological relevance, and safety of the top-ranked compounds.
The findings from this study provide computational evidence supporting the dengue virus NS5 polymerase (PDB ID: 2J7W) as a viable therapeutic target for drug repurposing. Several clinically approved antimalarial agents, including chloroquine, 3-hydroxyquinine, tetracycline, and artesunate, demonstrated favourable binding affinities and stable interactions with catalytically essential residues of the enzyme. These interactions suggest potential inhibitory effects on viral replication consistent with previous in vitro and in vivo observations.
Given their established safety, pharmacokinetic profiles, and global availability, these drugs represent promising scaffolds for further preclinical assessment and rational optimization against dengue virus infection. Future studies should integrate molecular dynamics simulations, enzymatic inhibition assays, and animal model testing to confirm their antiviral efficacy and clarify the molecular basis of NS5 inhibition. Despite the limitations associated with the co-crystallized ligand used for active-site mapping, the current results underscore the potential of structure-based repurposing strategies in accelerating antiviral drug discovery for dengue and related flaviviral diseases.
What is already known about the topic
What this study adds
A.A.A developed the protocol of the study, supervised data collection, data processing and analysis, developed and reviewed the manuscript. A.A.A., O.O., and E.A.A supervised data collection, participated in data processing and analysis, participated in manuscript development and review. A.A.A., O.O., and E.A.A facilitated data collection and compilation. A.A.A., O.O., and E.A.A participated in protocol and manuscript preparation and review. All authors read and approved the final manuscript.
Supplementary Files
| Table 1: Docking and MMGBSA Scores for the Top Drug Candidates | ||
|---|---|---|
| Drug candidate | MMGBSA dG Bind (kcal/mol) | Docking score (kcal/mol) |
| Chloroquine | -40.66 | -4.109 |
| 3-hydroxyquinine | -36.95 | -4.01 |
| Tetracycline | -36.69 | -6.494 |
| Artesunate | -35.17 | -4.4 |
| Chlorproguanil | -34.18 | -4.021 |
| Doxycycline | -30.27 | -4.796 |
| Monodesethylamodiaquine | -29.33 | -4.115 |
| Proguanil | -29.23 | -4.022 |
| Clindamycin | -27.78 | -5.828 |
| Co-ligand | -12.02 | -6.191 |
| Table 2: Post docking analysis showing the H bond, Hydrophobic interactions, and other interactions for the top six drug candidates | |||
|---|---|---|---|
| Drug candidate | H bond | Hydrophobic interactions | Other interactions |
| Chloroquine | None | CYS 709, TRP 795, ILE 797, ALA 799, TYR 766, TRP 803, MET 765, MET 761, LEU 511 | Pi-pi stacking: HIS 711 |
| 3-hydroxyquinine | ARG 729, SER 796 | LEU 734, TYR 758, TYR 766, MET 761, TRP 795, LEU 511, ALA 799, CYS 709 | Pi-pi stacking: HIS 711 (2) |
| Tetracycline | ARG 729, CYS 709, SER 796 (2), ASP 664, SER 661 | MET 761, TYR 766, LEU 511, CYS 709, ILE 797, ALA 799, TYR 606 | None |
| Artesunate | TYR 766, TRP 795 | TRP 803, TYR 758, MET 761, MET 765, TYR 766, LEU 511, TRP 795, LEU 734 | None |
| Chlorproguanil | SER 710 | TRP 803, MET 761, TYR 766, LEU 511, ILE 797, ALA 799, CYS 709, TYR 606 | Pi-pi stacking: HIS 711 |
| Doxycycline | SER 661, SER 796 | TYR 766, MET 761, LEU 511, TRP 795, ILE 797, ALA 799, CYS 709, TYR 606 | None |